我室神经科学团队在Nature Communications(IF=17.3)发表重要成果:首次证实TDP-43 功能丧失通过异常剪接产生神经毒性肽 PKN207,并在 AD 患者脑中检出,为神经退行性疾病诊疗提供新方向。
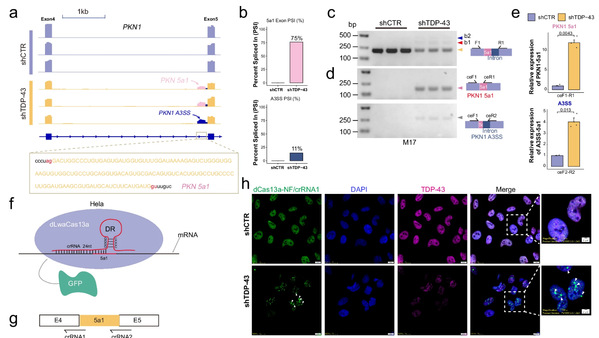
TDP-43 功能丧失是神经退行性疾病核心病理改变,可导致基因异常剪接,但能否产生稳定毒性蛋白尚不明确。本研究发现,TDP-43 缺失可特异性激活 PKN1 基因隐性外显子 PKN1-5a1,其转录本逃逸降解并翻译生成稳定截短肽 PKN207。该肽段仅在伴 TDP-43 病理的 AD 患者脑中检出,与疾病状态直接相关。动物实验显示,PKN207 在小鼠海马区过表达可造成认知记忆障碍与突触可塑性损伤,并通过抑制突触通路、扰乱细胞骨架稳态发挥神经毒性。
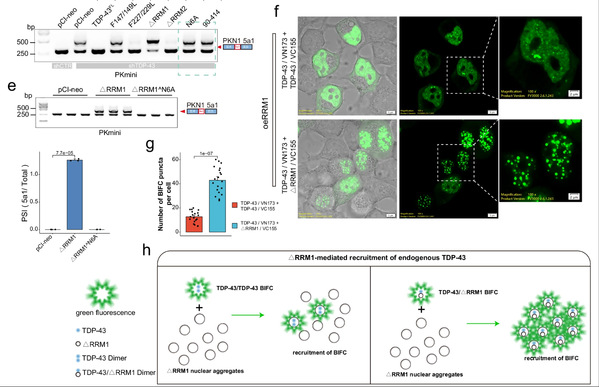
本研究首次系统地证实,TDP-43 功能丧失不仅通过 “功能丧失”机制 致病,还可通过“毒性获得” 新机制,即产生稳定毒性肽 PKN207 驱动神经退行病变。这一发现扩展了人们对TDP-43蛋白病发病机制的理解,将致病载体从传统的“错误折叠蛋白聚集”延伸至“异常剪接产生的稳定毒性肽”。PKN207作为一种在患者脑内可检测、在动物模型中验证具有神经毒性的分子,具备成为新型生物标志物和治疗靶点的潜力。
(文章链接https://doi.org/10.1038/s41467-026-68916-0)